PZLASTの使い方
(Go to English page)
1. ジョブの登録

-
PZLASTは、ユーザがアップロードしたタンパク質配列を、数テラバイトを超えるサイズの公共メタゲノムデータに対して検索し、
それらの自然環境中(あるいは人体組織中)の分布を調べることができるウェブアプリケーションです。
- 入力ファイル
- FASTA形式の**タンパク質**配列ファイルのみを入力として受け付けます。(DNA配列には対応していません) シングルFASTA形式、マルチFASTA形式のどちらでもOKです。 入力ボックス(B)に配列をペーストするか、ボタン(C)をクリックしてFASTAファイルを選択してください。
- パラメータ
- PZLASTは以下のパラメータによって検索機能をコントロールできます。
- 検索モード(D)
- 「環境検索」モードにより検索対象環境を限定するか、「シングルサンプル検索」モードにより特定の1サンプルのみを対象に検索ができます。デフォルト設定では、すべての環境が検索対象となります。
- "Output top N hits" (E)
- 入力配列ごとに上位何個までのヒットを出力するかを制御するパラメータです。トップヒットのみ(出力1)に設定することもできますが、大きめの値の方が面白い結果が得られやすいと思います。
- E-value cut-off (F)
- E-valueのカットオフ値を設定します。デフォルト値は1e-8です。この値を小さくすると、より類似性の高い配列に検索結果を限定できますが、ヒット数が減少する可能性があります。
- サブミットボタン(G)を押すとジョブが登録されます。
- 入力ファイルの制限
- すべての入力配列が10 AA (アミノ酸残基) 以上 2,000 AA以下であること。入力配列の総数が10,000本以下であること。 すべての配列の長さの総和が100,000AA以下であること。可能な出力ヒット数が100万以下であること。 したがってシングルFASTAを入力した場合は"Output top N hits"を100万に設定できますが、2本以上を入力した場合は、両方の配列について100万ヒットの出力を得ることはできません。
トップページ、またはナビゲーションバーの"Query"(A)をクリックして左画面へ移動してください。
-
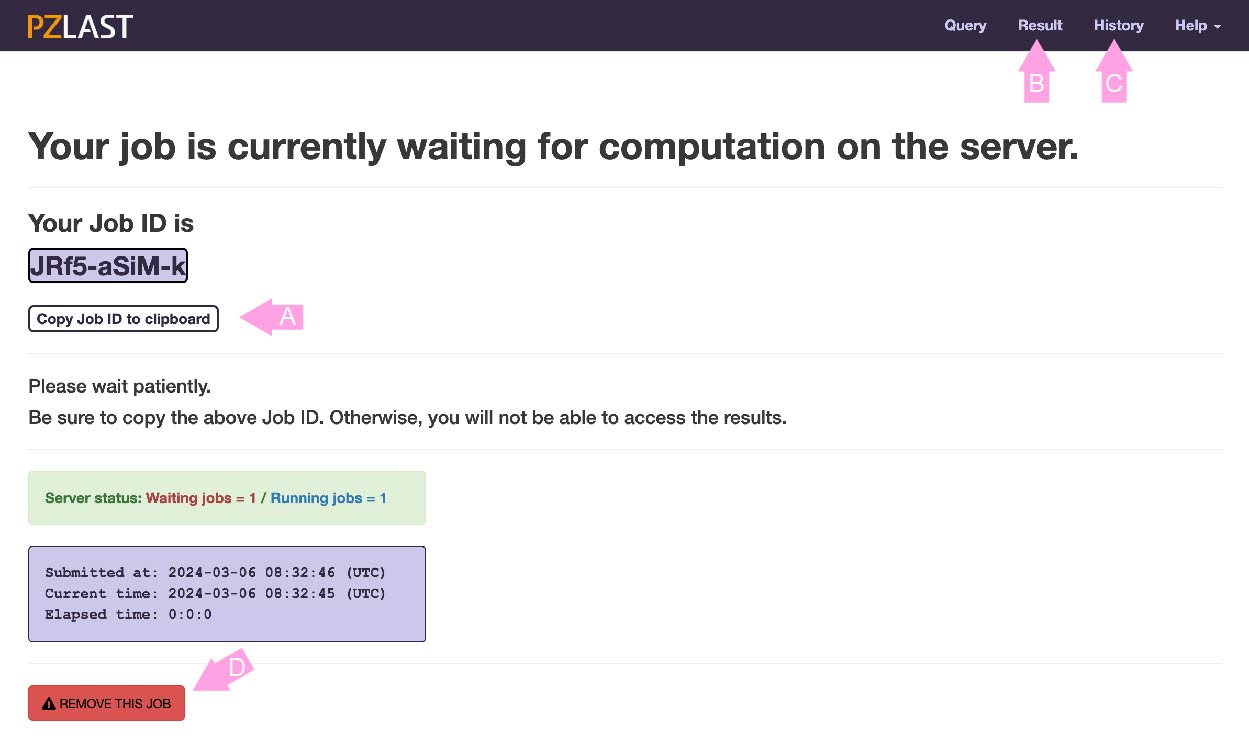
ジョブ登録ボタンを押すと、ジョブステータス表示ページに遷移します。
このページでは登録したジョブが現在、実行待ち状態なのか、 ZettaScaler-3.0上で計算実行中なのかが表示されます。 このページは数秒ごとに更新されます。 ZettaScaler-3.0上での計算が終了した場合(あるいは残念ながらエラーまたはヒット無しで終了した場合)、このページは結果ページへ自動的に遷移します。
すべてのジョブは、登録時に発行される固有のジョブIDで管理されます。
このページを離れる場合は、コピーボタン(A)をクリックしてジョブIDをクリップボード(あるいはどこか適当な場所)にコピーしておいてください。 ナビゲーションバーの"Result"(B)をクリックしてジョブIDを入力すれば、このページに戻ってくることができます。 また、"History"ページ(C)から過去のジョブを遡ることもできます。ただし、ジョブは登録後二週間で削除されるので注意してください。
※このウェブサイトではユーザのジョブ履歴を記録するためにCookieを使用しています。 ジョブIDと登録時間のみをCookieに記録しています。 Historyが表示されない場合は、ブラウザでCookieを有効にしてください。
実行待ち状態のジョブを削除したい場合は、"REMOVE THIS JOB"ボタン(D)を押してください。
2. 検索結果ページ
- Info
-
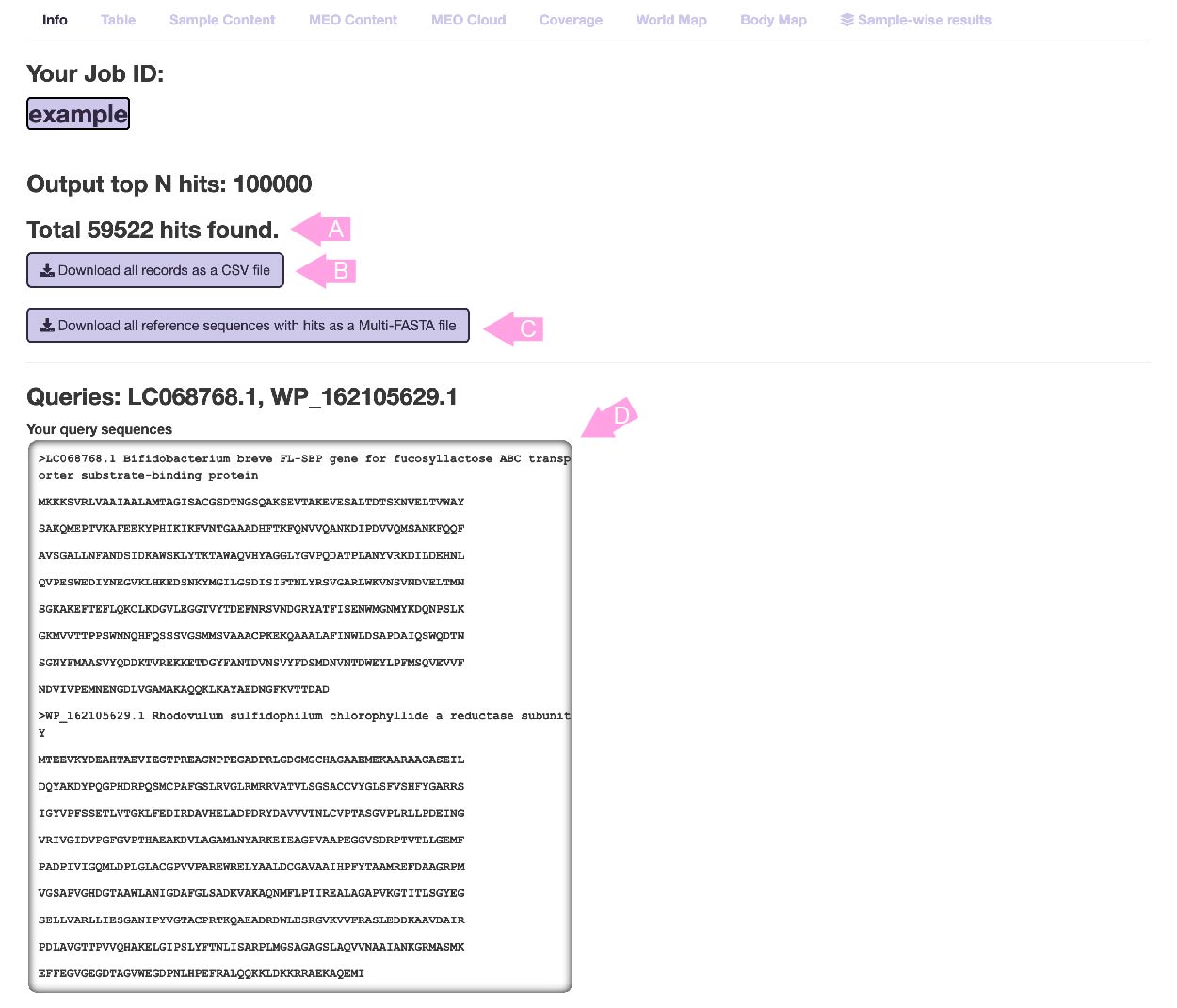
計算が完了すると、自動的にこのページが表示されます。
トータルのヒット数(マルチFASTAを入力した場合はそれぞれのヒット数の和)が(A)に表示されます。
(B)のボタンを押すと、すべての結果をCSV形式でダウンロードできます。
(C)のボタンを押すと、ヒットしたリファレンス配列のすべてをMulti-FASTA形式でダウンロードできます。
また、入力した配列の情報が(D)に表示されます。 結果はすべて"Queries"に表示されている配列IDで区別されるので、以下の結果ページをチェックする際はこれらのIDを覚えていてください。
- Table
-
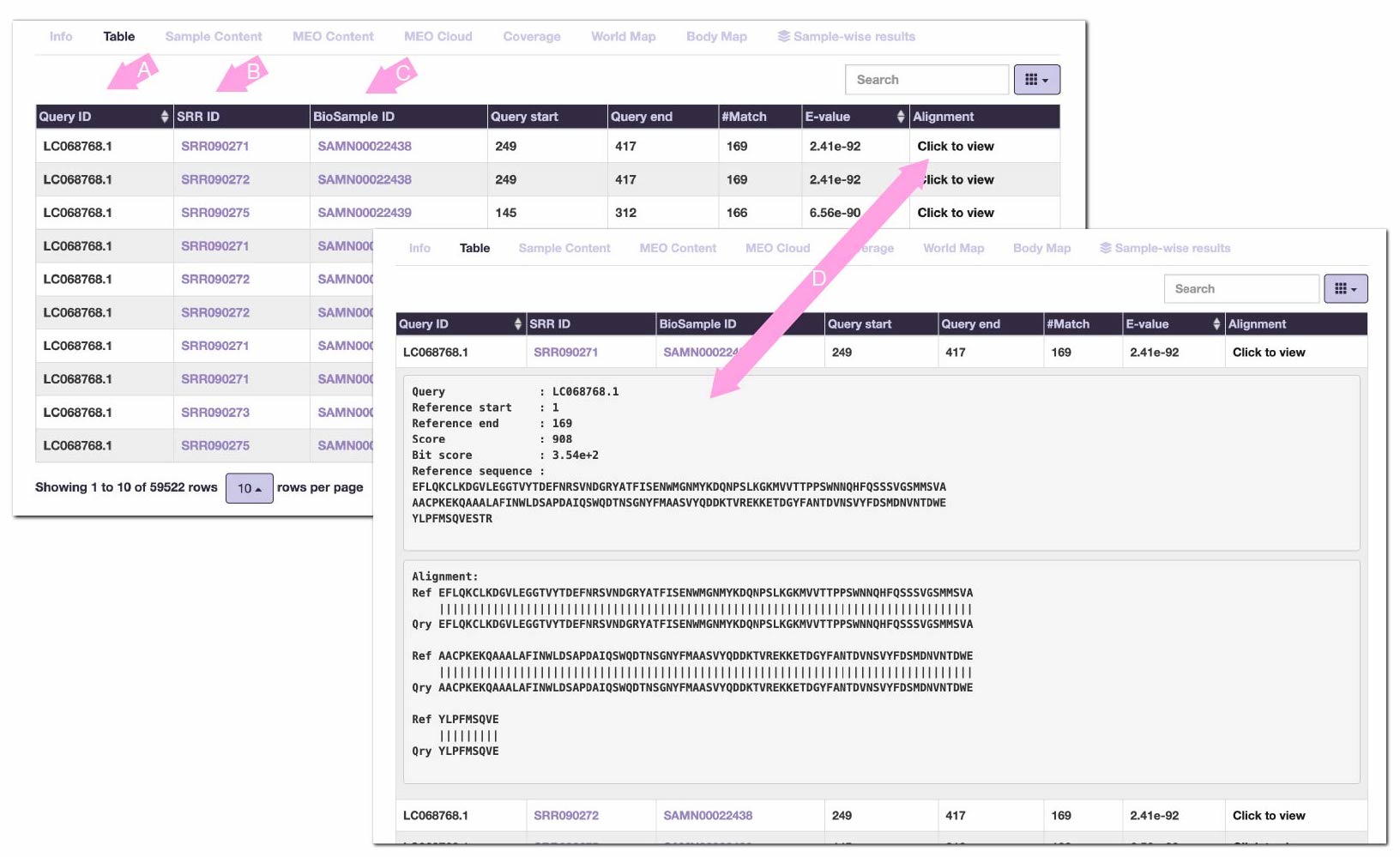
検索結果は、BLASTなどのツールと同様の表形式で表示されます。
デフォルトでは、ヒットレコードは入力配列ごとにE-valueの昇順で並んでいます。
このテーブルは「どのメタゲノムサンプルに含まれる配列にヒットしたか」を表示します。
(A)は入力配列のIDです。この列のヘッダをクリックすると、入力配列IDでテーブルをソートできます。
(B)はヒットした配列が含まれるメタゲノムサンプルのSRRアクセッション番号です。クリックするとNCBI SRA の該当ページに移動します。
(C)は対応するBioSample IDです。クリックするとNCBI BioSample の該当ページに移動します。
(D)をクリックすると、ヒットした参照タンパク質配列とアラインメントの詳細が個別のレコードごとに展開されます。
アラインメント中の文字は以下を意味します:
'|' ... マッチ
':' ... BLOSUM62スコア > 0
'.' ... BLOSUM62スコア = 0
'*' ... BLOSUM62スコア < 0

- Sample content
-
これ以降の結果ページでは、それぞれの**入力配列ごと**の結果が表示されます。
クエリを切り替える場合は、Aのボタンを押して結果を表示したいクエリを選択してください。
このページでは、各メタゲノムサンプルのヒット数を集計した結果を表示しています。 それぞれのサンプルに含まれる配列の何本にクエリがヒットしたかを棒グラフで表示しています。
サンプルのIDをクリック(B)するとNCBI BioSampleの該当ページにジャンプし、そのサンプルについての詳しい情報を得ることができます。
デフォルトではすべてのヒットについて集計していますが、集計の範囲を上位のヒットのみに限定する(C)ことも可能です。
さらに、ヒットした本数そのものではなく、各サンプルの総配列本数に対するヒットした配列の割合(%)の表示に切り替えることも可能です。(D)(E)

- MEO content
-
MicrobeDB.jpでは、すべてのメタゲノムサンプルはMEO (Metagenome and Microbes Environmental Ontology)によってアノテーションされています。
MEOは、そのサンプルがどのような自然環境あるいはヒト共生環境から取得されたのかを記述する統一的なオントロジーです。
この棒グラフは、あるMEOに紐づいた何個のサンプルにクエリがヒットしたかを表示しています。(ヒット数ではなくサンプル数です。1ヒットでも見つかったサンプルはカウントされます)
また、"Sample content"のページと同様、そのMEOを持っているサンプルのうちどれだけの割合(%)のサンプルにヒットしたかの表示に切り替えることも可能です。(B)(C)
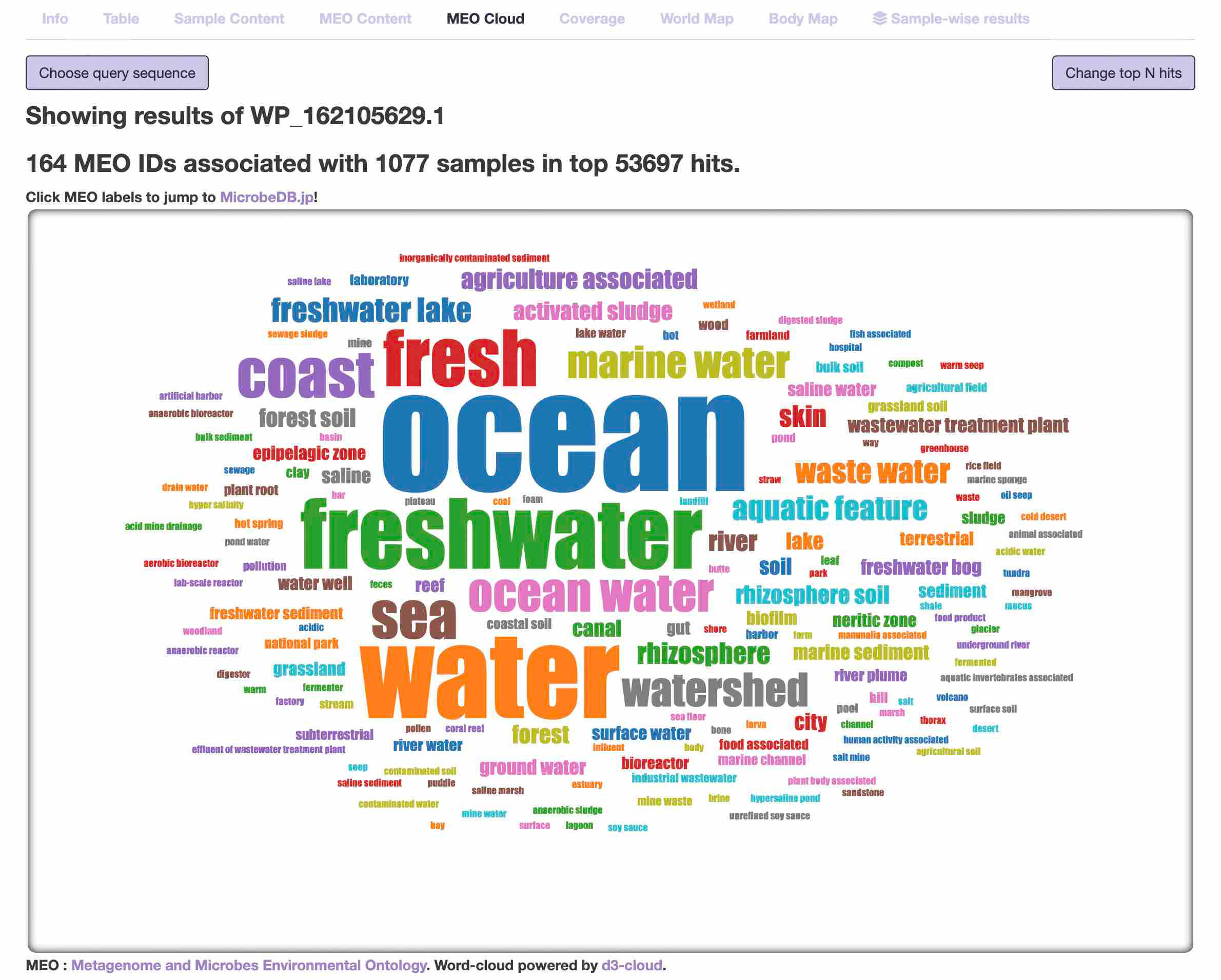
- MEO cloud
-
本質的に"MEO content"で可視化されている情報とまったく同じですが、ワードクラウドでちょっとオシャレに表示されています。
MEO語彙のサイズは、ヒットサンプルの数に比例してスケールしています。
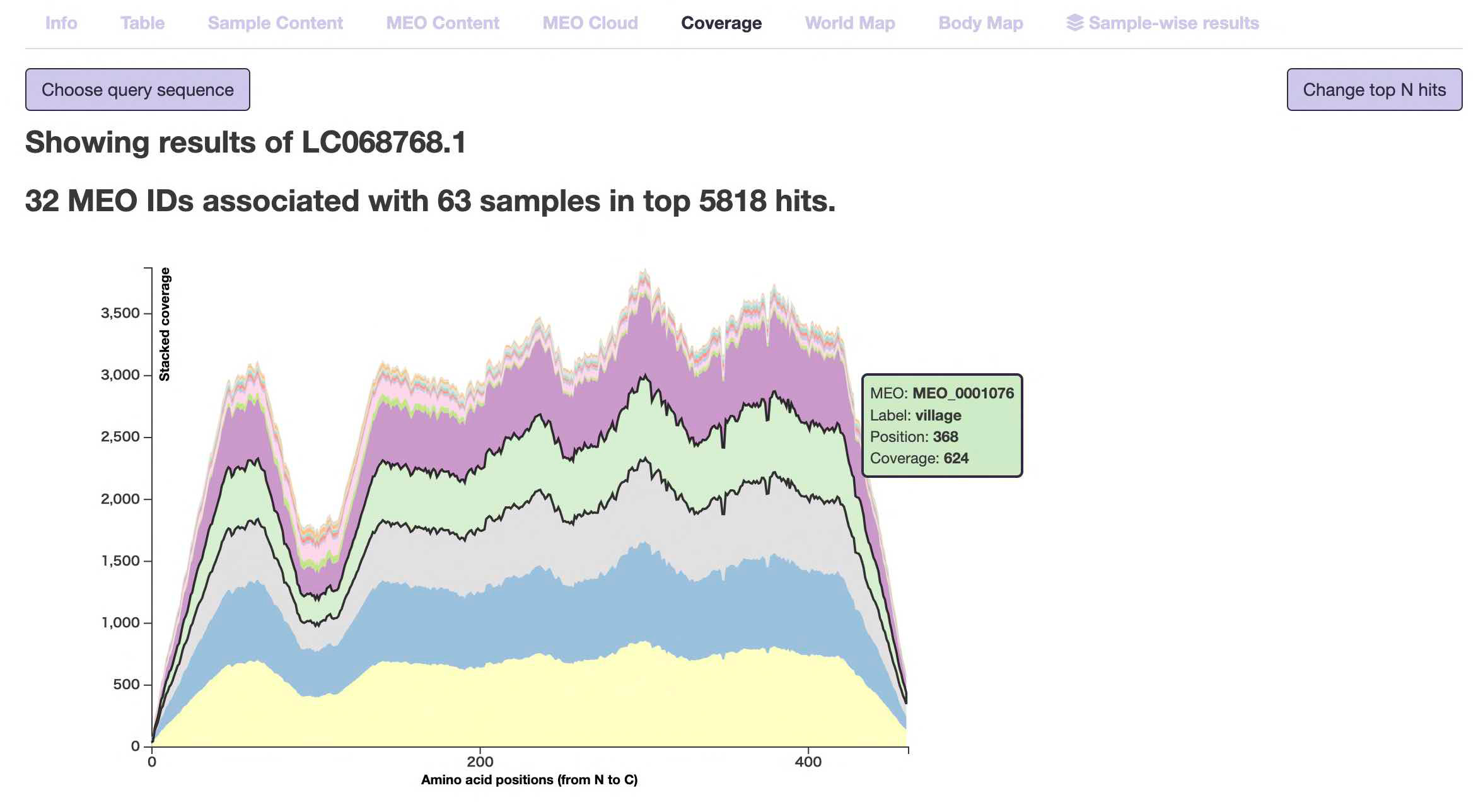
- Coverage
-
入力されたタンパク質クエリのN末端からC末端にかけて、各アミノ酸の位置における「環境カバレッジ」を表示します。これにより、特定のドメインが特定の環境にのみ存在するといった関係性を発見することが可能になります。
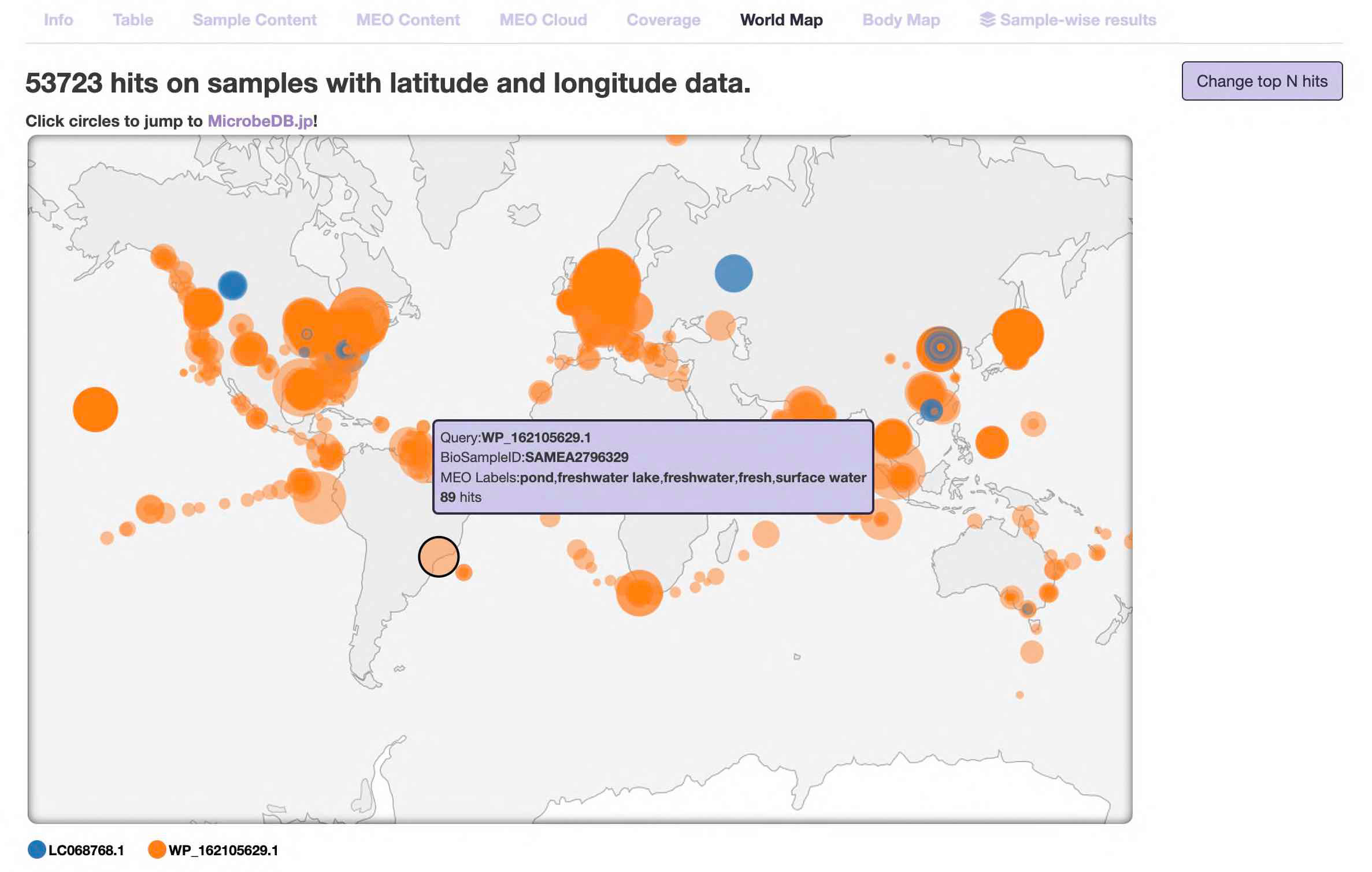
- World map
-
ヒットしたサンプルの「場所」を世界地図上に表示します。
注1:ヒットサンプルのうち、このチャートには緯度と経度の情報が登録されているサンプルのみが表示されます。
注2:緯度と経度の情報は、公共データベースに登録するときに各研究者によって記述されたものです。必ずしもサンプルがどこで採取されたか、被験者がどこに住んでいるかを示しているとは限りません。
ヒットした配列数が多いサンプルほど、大きな円で表示されています。
このページに関しては、複数のクエリの情報が同時に表示されています。 異なる色の円は異なるクエリに対応しています(左下の凡例を参照)。
円をマウスオーバーすると、サンプルID、MEOラベルなどの情報が表示されます。
円をクリックすると、NCBI BioSampleの該当サンプルページにジャンプします。
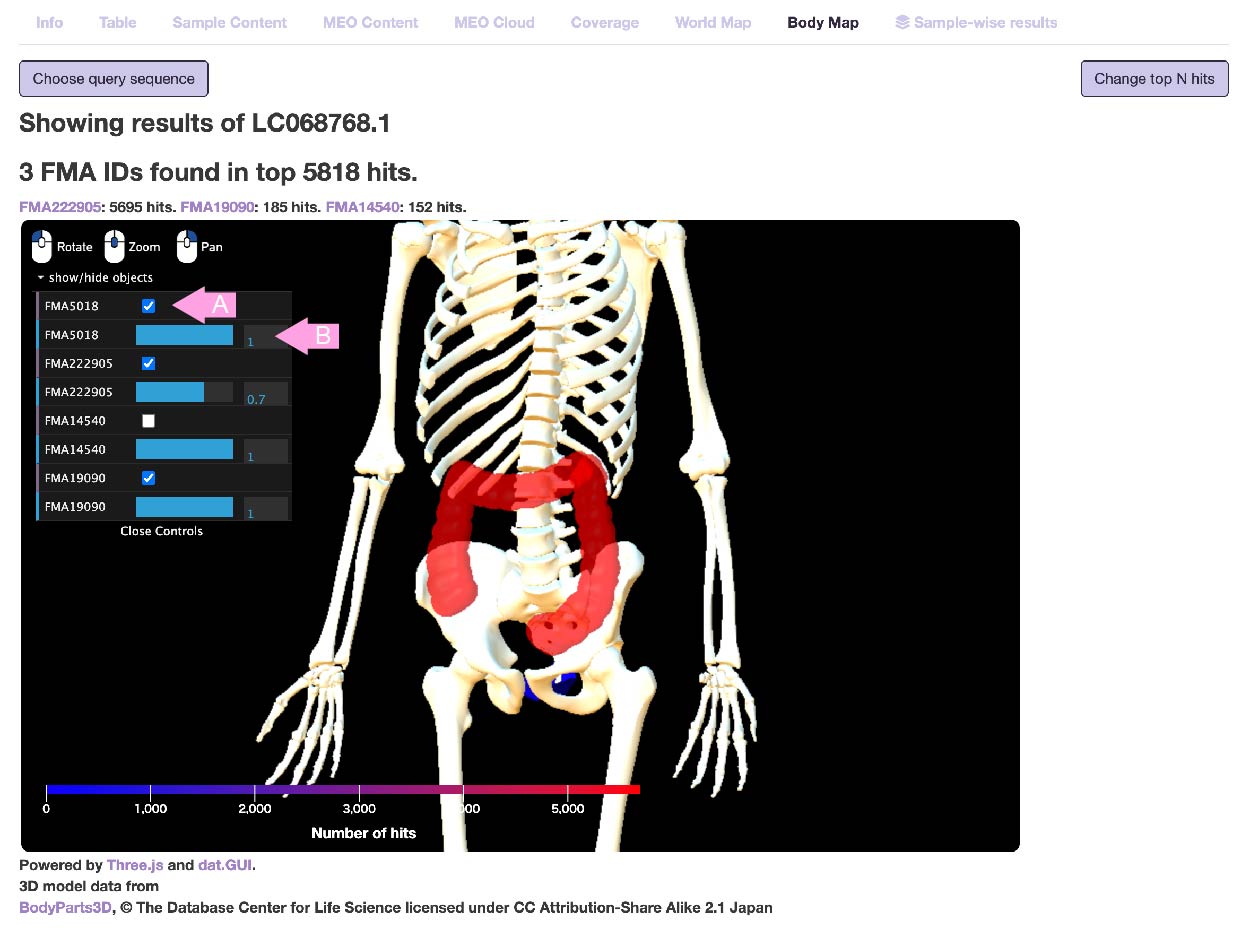
- Body map
-
ヒトに由来するメタゲノムサンプルの中には、FMA (Foundational Model of Anatomy Ontology)のIDを持つサンプルがあります。
ここでは、クエリがヒットしたサンプルのうち、ヒト共生環境に由来するサンプルについて、BodyParts3D の臓器3次元モデルを利用して表示しています。
注:必ずしも表示されている臓器そのものを対象としたメタゲノムサンプルにヒットしたとは限りません。ヒトの糞便サンプルはしばしば大腸と関連づけられています。
その臓器に関連したサンプルの配列ヒット数が多いほど、その臓器の色が赤く表示されます。
骨だけは常に表示されますが、ヒットしなかった他の臓器は表示されません。
左クリックで回転、右クリックで平行移動、マウスホイールで拡大縮小できます。
左側のコントロールパネルを使って、表示されている臓器それぞれについて、表示・非表示の切り替え(A)と透明度の調整(B)が可能です。
3. サンプルごとの集計結果 (Experimental)

- Completion table
-
以降のページでは、クエリ配列が特定サンプルの特定配列にヒットした個々の結果ではなく、各メタゲノムサンプルごとの合計ヒット数に基づく集計結果を表示します。
(A)をクリックすると、"Sample-wise results" に関連する結果ページが表示されます。
このページでは、Multi-FASTA形式で入力された複数のタンパク質配列の充足率に基づき、ヒットしたメタゲノムサンプルをソートして表示します。
充足率とは、入力された複数のクエリタンパク質配列が同一のメタゲノムサンプル内で見つかる割合を指し、一配列でもヒットした時点で「見つかった」と判断されます。
この機能により、例えば、特定の代謝パスウェイに関与する複数の酵素タンパク質配列を一括して検索し、それらが揃って存在するメタゲノムサンプルを特定することが可能になります。
(B)は各サンプルにおけるクエリタンパク質の存在または不在を示しています。(C)は、各サンプルのCompletion ratio(充足率)を表しています。

- Completion-MEO Cloud
-
充足率が高い環境をワードクラウドで表示します。
MEO語彙のサイズは充足率に比例してスケールされます。
まず、各メタゲノムサンプルに充足率に基づいて重み付けを行い、次にそのサンプルが持つMEO語彙に対する加重和を計算します。
これによってMEO語彙のサイズが決定され、大きく表示される環境は、入力された配列が同時に観測される可能性が高い環境であることを示します。
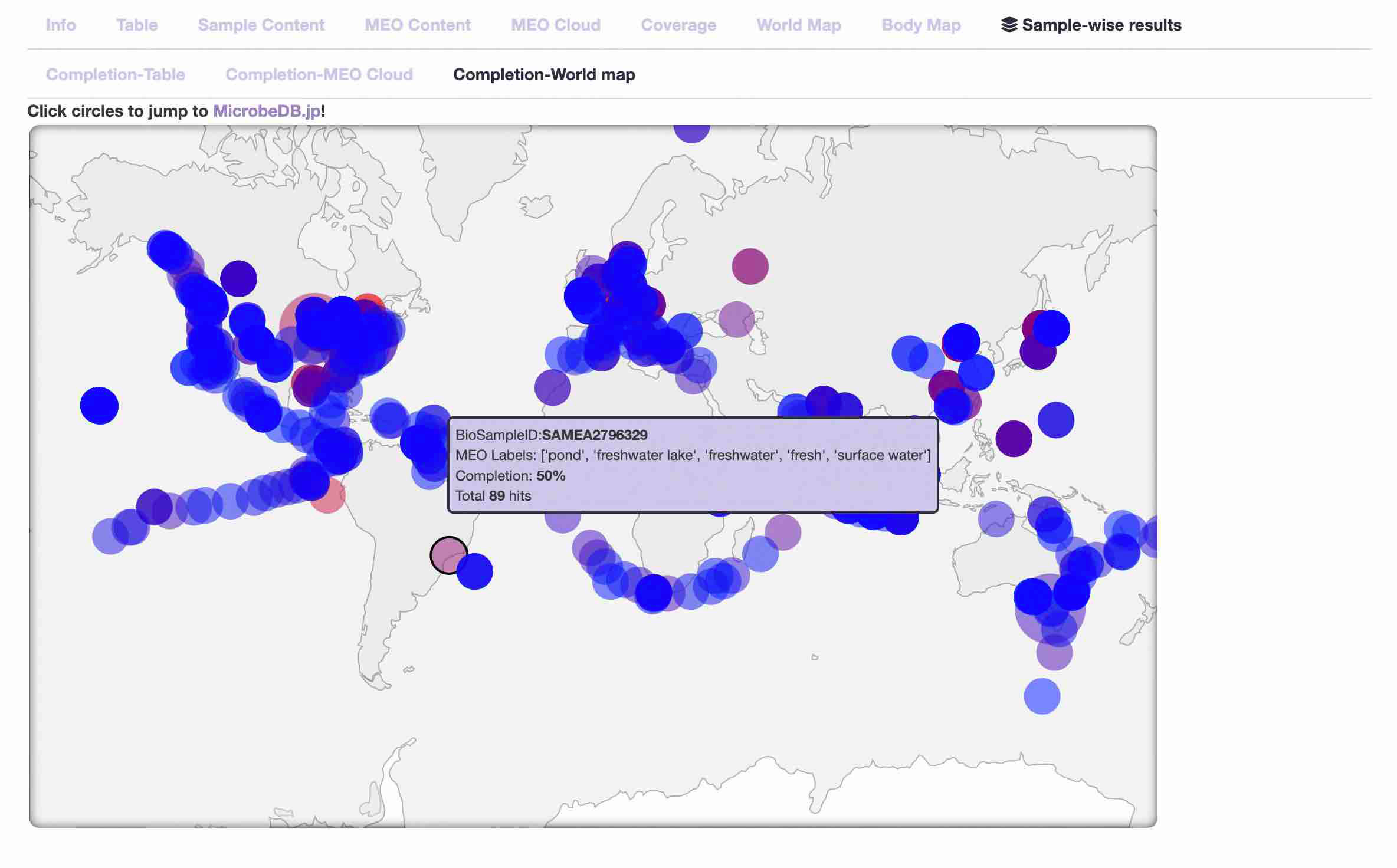
- Completion-World map
-
充足率が高いサンプルの場所を世界地図上に表示します。
円のサイズが大きいほど充足率が高いサンプルであることを示し、円の色が赤色に近いほどクエリタンパク質の平均ヒット数が多いことを表しています。